Korrigerende sikkerhetstiltak for medisinsk utstyr
Publisert:
|
Oppdatert:
Endringer
Produsenter, autoriserte representanter, importører og distributører har en lovpålagt plikt til å rapportere eller videreformidle korrigerende sikkerhetstiltak.
Innhold på siden
Hva er et korrigerende sikkerhetstiltak
Korrigerende sikkerhetstiltak er et tiltak produsenter kan iverksette basert på tekniske eller medisinske årsaker for å forhindre eller redusere risikoen for alvorlige hendelser ved bruk at et medisinsk utstyr som er satt på markedet.
Ved usikkerhet om egen rolle se informasjon om ulike roller for markedsaktører.
Ved funn av systematiske feil eller mangler eller andre hensyn som påvirker utstyrets sikkerhet i bruk skal produsenten iverksette korrigerende sikkerhetstiltak (Field Safety Corrective Action – FSCA). Hensikten kan være å utbedre feil, informere brukere om riktig og sikker bruk og minimere risikoen for alvorlige hendelser ved bruk av utstyret. Det korrigerende sikkerhetstiltaket varsles brukere ved hjelp av en sikkerhetsmelding (Field safety notice – FSN).
Eksempler på korrigerende sikkerhetstiltak
-
Tilbaketrekking av utstyret fra markedet
-
Endringer på utstyr, midlertidige eller permanente, eks delebytte eller bruksanvisning, softwareoppdateringer
-
Informasjon om endringer i vedlikehold
-
Anbefalinger om bruken av utstyret, kalibreringsbehov eller i henhold til analyse av pasientresultater.
-
Endringer i oppbevaring av prøver eller reagens
Produsenters og autorisert representanters (AR) ansvar
Produsent skal vurdere behov for sikkerhetstiltak ut ifra sitt risikostyringssystem. Produsent og AR plikter å rapportere korrigerende sikkerhetstiltak til ansvarlig myndighet i det landet der produsenten eller AR har sin forretningsadresse. Norske produsenter skal derfor rapportere sine FSCA til DMP.
Rapport skal også sendes til myndighetene i samtlige berørte land i EU/EØS der det aktuelle utstyret er på markedet. Dersom utstyret er sertifisert av et meldt organ, skal også det meldte organet underrettes.
Produsenten skal ved hjelp av en sikkerhetsmelding sørge for at brukerne av det aktuelle utstyret underrettes om det korrigerende sikkerhetstiltaket som er truffet.
Hvordan rapportere et korrigerende sikkerhetstiltak
Importørers og Distributørers ansvar
Importører og distributører skal samarbeide med produsenten for å sikre at det treffes nødvendige korrigerende sikkerhetstiltak for utstyr de har omsatt. Dersom det er relevant, kan samarbeidet også inkludere AR og ansvarlig myndigheter.
Importører og distributører skal:
-
Se til at det korrigerende sikkerhetstiltaket gjennomføres i henhold til produsentens plan
-
Distribuere produsentens sikkerhetsmeldinger til sine brukere eller neste ledd i omsetningskjeden
-
Holde oversikt over tiltakets gjennomføring for sitt marked
-
Føre register over utstyr som omfattes av sikkerhetsmeldinger
-
Underrette produsent (og evnt AR) dersom de vurderer eller har grunn til å tro at utstyr de har brakt i omsetning har feil eller mangler som påvirker utstyrets sikkerhet
-
Underrette produsent dersom de mottar klager eller rapporter fra helsepersonell
Helsevirksomhetens ansvar (Brukere og eiere)
Sikkerhetsmeldingen beskriver hvordan brukere og eiere skal forholde seg til sikkerhetstiltaket og hvordan informasjonen evt. skal videreformidles.
Finn mer om håndtering av sikkerhetsmeldinger på egen temaside.
Privatpersoner
Dersom du som privatpersoner blir kjent med et sikkerhetstiltak som angår utstyr du benytter. Forhold deg til instruksene som blir gitt, alternativt ta kontakt med aktøren som utleverte utstyret.
Oversikt over de ulike rollene for markedsdeltakere
| Markedsdeltaker | Definisjon i MDR artikkel 2 og IVDR artikkel 2 |
|---|---|
| Produsent | en fysisk eller juridisk person som framstiller eller helrenoverer utstyr, eller som får utstyr designet, framstilt eller helrenovert, og som markedsfører nevnte utstyr i eget navn eller under eget varemerke. |
| Autorisert representant | enhver fysisk eller juridisk person etablert i Unionen* som har fått og har akseptert en skriftlig fullmakt fra en produsent plassert utenfor Unionen, til å utføre bestemte oppgaver på dennes vegne med hensyn til dennes forpliktelser i henhold til denne forordning. |
| Importør | enhver fysisk eller juridisk person etablert i Unionen* som bringer utstyr fra en tredjestat i omsetning i Unionen*. |
| Distributør | enhver fysisk eller juridisk person i omsetningskjeden, utenom produsenten eller importøren, som gjør utstyr tilgjengelig på markedet fram til ibruktaking. |
| Helseinstitusjon | en organisasjon som har som hovedformål å pleie eller behandle pasienter eller fremme folkehelsen. |
* Med «Unionen» menes her EU/EØS-området og Tyrkia.
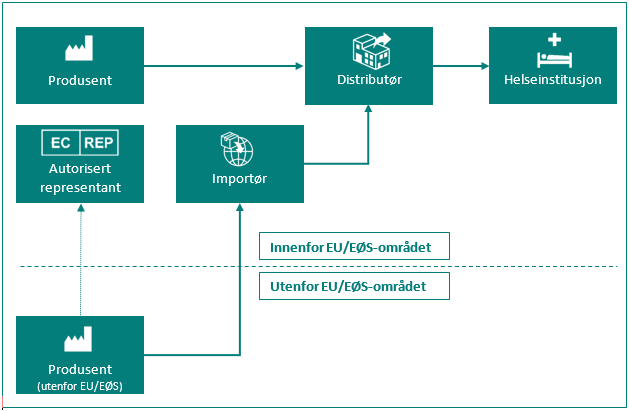
Figur: Markedsdeltaker i omsetningskjeden.
DMPs rolle ved et sikkerhetstiltak
Det er produsentens ansvar at det korrigerende sikkerhetstiltaket gjennomføres som planlagt. DMPs oppgave er å vurdere mottatt rapport over sikkerhetstiltaket og overvåke at det blir gjennomført som planlagt. DMP vurderer om produsentens korrigerende sikkerhetstiltak er tilstrekkelig, og om det kan være behov for endringer eller ytterligere korrigerende tiltak.
DMP samarbeider med andre myndigheter i EU om å overvåke sikkerhetstiltak. Når en norsk produsent rapporterer et korrigerende sikkerhetstiltak til DMP, deles dette med det europeiske nettverket slik at andre myndighetene der utstyret er på markedet blir informert. Dette er i tillegg til produsentens egen varsling til berørte myndigheter.